Phenylketonuria Research Services
Phenylketonuria is an autosomal recessive disorder caused mainly by defects in phenylalanine hydroxylase (PAH) or its cofactor tetrahydrobiopterin (BH4), resulting in the inability to metabolize phenylalanine to tyrosine properly, which accumulates in the body and triggers a series of pathologic changes
Causes of Phenylketonuria
Reduced or complete loss of PAH activity in the liver can result in phenylalanine not being properly converted to tyrosine, which accumulates and is metabolized in the body to substances such as phenylpyruvic acid, phenylacetic acid, and phenyllactic acid, which are excreted through the urine, forming the typical features of phenylketonuria. In addition, BH4 deficiency leads to dysfunction of the enzyme phenylalanine hydroxylase, further exacerbating the abnormal metabolism of phenylalanine.
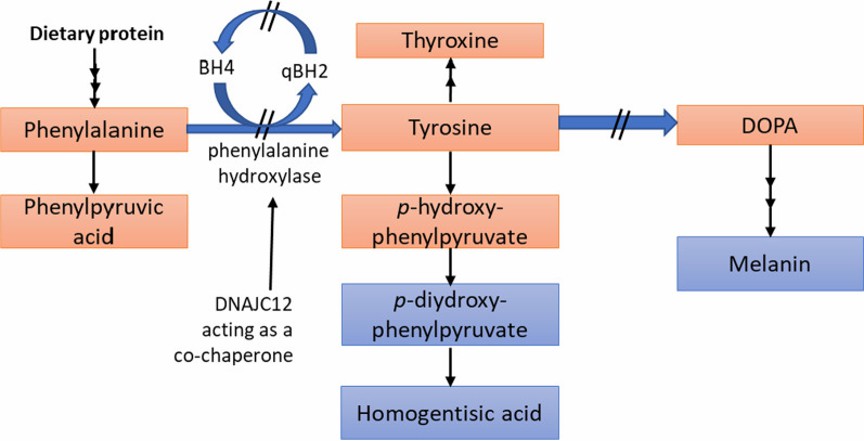 Figure 1. Phenylalanine metabolism in Phenylketonuria. (Elhawary NA, et al., 2022)
Figure 1. Phenylalanine metabolism in Phenylketonuria. (Elhawary NA, et al., 2022)Treatment of Phenylketonuria
- Dietary therapy
- Drug therapy: BH4 supplements
- Gene therapy
- Probiotic therapy
Principle of Phenylketonuria Gene Therapy
By introducing a functional PAH gene into the patient in order to restore the normal metabolic function of phenylalanine hydroxylase. For example, NGGT002 is a recombinant AAV vector gene therapy drug that drives PAH gene expression through a liver-specific promoter. This drug has shown safety and efficacy in phase I/II clinical trials and is expected to be a long-term treatment for phenylketonuria.
Disclaimer: Protheragen specializes in providing preclinical research services. The above is for informational purposes only. It is not intended as a recommendation for a treatment program. For guidance on treatment options, please visit the regular hospital.
Our Services
Diagnostic Method Development Services for Phenylketonuria
Types of diagnostic methods we support:
- Tandem mass spectrometry (MS/MS)
- Gene sequencing technologies: PCR-STR, ASPCR, PCR-SSCP, Sanger sequencing
- Fluorescence detection method
Our advanced gene sequencing technology not only accurately identifies mutation loci, but also provides an important basis for prenatal diagnosis. We are committed to providing strong support for early screening, confirmation of diagnosis and treatment of phenylketonuria.
Model Development Services for Phenylketonuria
In Vitro Models
- K562-D cell model
- Human induced pluripotent stem cells (hiPSCs) engineered organ model
In Vivo Models
- Pahenu2/Enuhu2 mouse model
- Gene editing mouse model
Our in vitro model can be used to analyze the specific mechanisms of phenylalanine metabolism. Our in vivo model is closer to the real physiological environment and can fully reflect the pathological process of PKU and its effects on the nervous system.
Metabolic Analysis of Phenylketonuria
- Metabolic pathway analysis
- Metabolite analysis, e.g. phenylpyruvic acid, phenylacetic acid, alpha-ketoglutaric acid
- Metabolic pathway bypass analysis
We are committed to providing important support for the early detection, diagnosis and treatment of phenylketonuria through a variety of metabolic analysis methods.
Therapy Development Services for Phenylketonuria
The types of therapy development we support:
- Engineered bacteria therapy, genetically engineered microorganisms to achieve effective degradation of phenylalanine.
- Gene replacement therapy, gene editing therapy
Additional Services:
- Further optimization of vector design, improvement of therapeutic efficiency and reduction of side effects.
- Developing safer and more efficient viral vectors
- Explore multiple delivery routes
Protheragen actively seeks partnerships with academic institutions and pharmaceutical companies to accelerate the development of innovative treatments for phenylketonuria. If you are interested in our services, please feel free to contact us.
Reference
- Elhawary NA, AlJahdali IA, Abumansour IS, et al. Genetic etiology and clinical challenges of phenylketonuria. Hum Genomics. 2022;16(1):22.
All of our services and products are intended for preclinical research use only and cannot be used to diagnose, treat or manage patients.