Clouston syndrome, also referred to as Hidrotic Ectodermal Dysplasia 2 (HED2), is a rare genetic condition resulting in abnormalities of the skin, hair, and nails as a result of mutations in the GJB6 gene. Protheragen provides a comprehensive series of preclinical services in drug and therapy development that is specialized to meet the challenges of Clouston syndrome and expedite the breakthroughs of your research project.
Introduction to Clouston Syndrome
Clouston syndrome is a genetic condition caused by mutations in the GJB6 gene that encodes the connexin 30 protein, which is important for cell communication of the skin and its appendages. Patients with Clouston Syndrome are typically affected by several manifestations including alopecia, nail dystrophy, and palmoplantar hyperkeratosis. However, while the prevalence is unknown, the condition is considered to be ultra-rare, creating substantial challenges for diagnosis and therapeutic development.
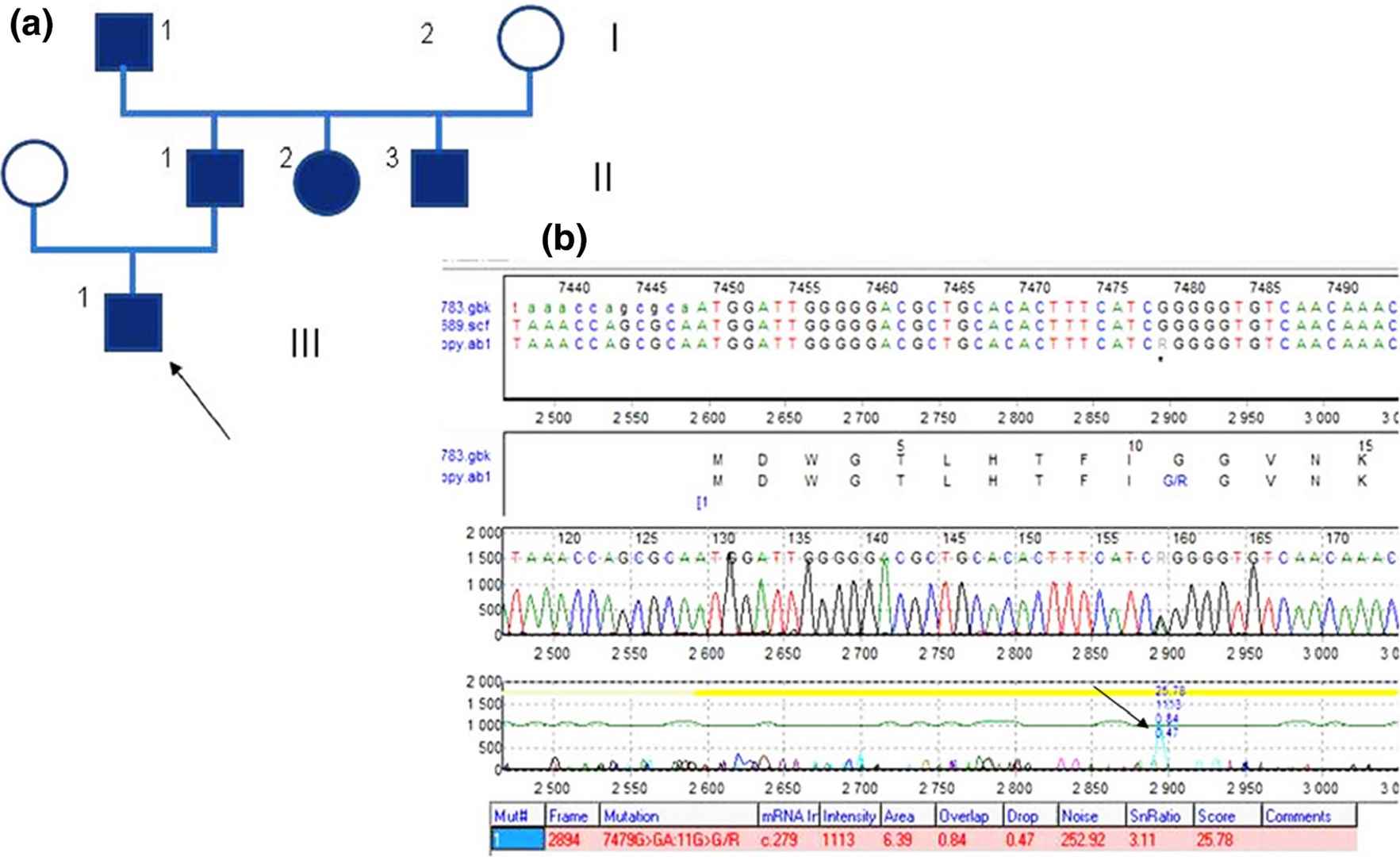
Fig.1 Pedigree of the Polish Clouston family and genetic results of the exon 1 the GJB6 gene analysis. (Pietrzak
et al., 2016)
Pathogenesis of Clouston Syndrome
Clouston syndrome is one of the forms of hidrotic ectodermal dysplasia, and is caused by mutations in the GJB6 gene, which encodes connexin 30, a gap junction protein critical for intercellular communication in ectodermal tissues. The mutations lead to dysfunctional connexin 30 channels, perturbing the formation of gap junctions and intercellular exchange of ions and small molecules. Recent evidence demonstrates that altered connexin 30 functions changes the proliferation and differentiation of keratinocytes and other ectodermal cells, creating the symptomatic abnormalities of hair, skin and nails seen in Clouston syndrome independent of sweat function.
Therapeutics Development for Clouston Syndrome
Gene Replacement Therapy
Delivering a functional copy of the GJB6 gene to affected cells, thereby restoring the normal expression of Connexin 30. The use of adeno-associated virus (AAV) vectors is very common and has an acceptable safety profile with efficiency. Recent studies have shown successful transduction of keratinocytes with wild-type GJB6, leading to restored gap junction functionality
Allele-Specific Silencing
Application of antisense oligonucleotides (ASOs) or RNA interference (RNAi) to the mutant GJB6 allele while retaining the expression of the wild-type gene. The goal of this approach is to eliminate the dominant-negative activity of the mutant protein. In one, the researchers significantly reduced the abundance of the mutant mRNA from patient-derived cells, leading to restored intercellular communications.
Our Services
Protheragen provides a full suite of services to move therapeutics forward for Clouston syndrome. Our team of expert scientists, dermatologists, and geneticists deploys innovative technologies to facilitate progress, with both dedicated therapeutic development and disease model development services to support your project.
Therapeutic Development Platforms for Clouston Syndrome
At Protheragen, we provide integrated end-to-end solutions for small molecule, gene therapy, cell therapy, and biologic platforms to address the GJB6-induced Connexin 30 dysfunction associated with Clouston syndrome for epidermal gap junction communication and barrier restoration.
Disease Models Development for Clouston Syndrome
Protheragen advanced preclinical disease models, including 2D cell models, 3D skin models and animal models development services specifically designed to model disease-specific phenotypes to accelerate therapeutic development.
Therapeutic Development Platforms for Classic Ehlers-Danlos Syndrome
| 2D Cell Models & 3D Skin Models |
| Protheragen's research services aimed at Clouston syndrome create a base for advancing new effective therapies. We perform in vitro studies using 2D cell models and 3D skin models learn about the molecular pathology underlying Clouston syndrome. |
| Optional Models |
- GJB6-Mutant Primary Keratinocytes
- GJB6-KO Keratinocyte Lines
|
- GJB6-Mutant 3D Epidermal Equivalents
- GJB6-Mutant Epidermal Organoids
|
| Animal models |
| In vivo preclinical studies are an important component of our services. We utilize, such as genetically engineering models, to test the safety and efficacy of potential therapeutics for Clouston syndrome. These animal models will be well characterized and selected based on their ability to model the human pathology of Clouston syndrome. |
| Optional Models |
- Epidermal GJB66 Conditional KO Mice
- GJB6a/b Double KO Zebrafish
|
|
| Optional Species |
Mice, Rats, Non-human primates, Others |
As a one-stop preclinical research services provider, Protheragen dedicated to advancing therapies for rare skin diseases like Clouston syndrome. Our end-to-end capabilities allow you to discover your target and disease model to perform full and complete drug safety evaluation and DMPK services. If you are interested in our services, please feel free to contact us.
References
- Murshidi, R., and H. Al-Lala. "Clouston Syndrome: Report of a Jordanian Family with Gjb6 Gene Mutation." Case Rep Dermatol Med 2023 (2023): 5577379.
- Pietrzak, A., et al. "Immune System Disturbances in Clouston Syndrome." Int J Dermatol 55.5 (2016): e241-9.
All of our services and products are intended for preclinical research use
only and cannot be used to diagnose, treat or manage patients.

